HISTIOZYTOSE
Unter histiozytäre Erkrankungen sind viele unterschiedliche Erkrankungen zusammengefasst. Erfahren Sie hier mehr rund um die Diagnose und Behandlung von Histiozytosen.
Wer wir sind
Im Histiozytose-Netzwerk haben sich Kolleginnen und Kollegen organisiert, die sich mit diesen seltenen Erkrankungen beschäftigen. Da diese Art von Erkrankung alle Organe betreffen kann, haben wir viele Spezialistinnen und Spezialisten vereinigt, um eine optimale Betreuung unserer Patientinnen und Patienten zu gewährleisten.
Über uns
Im Histiozytose-Netzwerk haben sich Kolleginnen und Kollegen organisiert, die sich mit diesen seltenen Erkrankungen beschäftigen. Da diese Art von Erkrankung alle Organe betreffen kann, haben wir viele Spezialistinnen und Spezialisten vereinigt, um eine optimale Betreuung unserer Patientinnen und Patienten zu gewährleisten.
In einem regelmässig stattfinden „Histiozytose-Board“ werden die Fälle interdisziplinär besprochen und Handlungsempfehlungen ausgesprochen. Natürlich wird auch darüber hinaus immer der Kontakt untereinander gesucht und die Behandlung untereinander abgestimmt.
Am Universitätsspital Zürich sind hochqualifizierte Kolleginnen und Kollegen aus den Kliniken für Medizinische Onkologie und Hämatologie, Pneumologie, Dermatologie, Endokrinologie, Pathologie, Radiologie und Nuklearmedizin, sowie der Allgemeinen Inneren Medizin im Netzwerk organisiert.
Um diese seltene Erkrankung besser zu verstehen, haben wir 2020 eine Registerstudie für erwachsene Patientinnen und Patienten mit histiozytären Erkrankungen initiiert, die laufend neue Fälle aufnimmt. Wenn Sie hierzu Fragen haben oder an der Studie teilnehmen wollen, finden Sie weitere Informationen hier.
Neben der klinischen Versorgung steht für uns auch die Fort- und Weiterbildung im Vordergrund. Hierfür steht die regelmässige Teilnahme an Nationalen und Internationalen Konferenzen und Präsentation von Ergebnisse aus unserer Wissenschaftlichen Arbeit.
Übersicht Präsentationen
In diesem Rahmen wird im Oktober 2023 das 1. Schweizer Histiozytose-Symposium stattfinden. Nähere Informationen zur Veranstaltung finden Sie hier.
Gerade in der Betreuung von Patientinnen und Patienten mit seltenen Erkrankungen ist ein Austausch auch mit internationalen Expertinnen und Experten wichtig. Wir sind daher Mitglied bei ECHO (European Consortium für Histiocytosis), sowie der ECD-Alliance und der Histiocytosis Society.
In der gesamten Schweiz haben wir Ansprechpartnerinnen und Ansprechpartner >
Ansprechpartner: innen CH.
Patientenforum Histiozytose
Oft ist die Diagnose einer histiozytären Erkrankung ein Schock. Nicht selten hat es lange gedauert, bis die Diagnose gestellt werden konnte und viele belastende Untersuchungen waren nötig. Aber was genau sind das für Erkrankungen? Und wie geht es weiter? Ist eine Therapie notwendig? Am Ende sogar eine Chemotherapie? Aber es ist doch kein Krebs, oder? „Das einzige, was mir mein Arzt sagen konnte ist, dass es eine seltene Erkrankung ist.“ So oder ähnlich berichten uns viele Patientinnen und Patienten, wenn sie zum ersten Mal zu uns in die Sprechstunden kommen. Mit unserem Angebot möchten wir Ihnen zeigen, dass Sie in dieser Situation nicht alleine sind. Spezialistinnen und Spezialisten aus allen Fachrichtungen sind hier versammelt, um Sie optimal zu beraten und, wenn nötig, die beste Behandlung auszuwählen. Wir möchten Ihnen wenn Sie es wünschen auch die Möglichkeit geben, sich mit anderen Betroffenen auszutauschen. Gerade mit einer so seltenen Erkrankung fühlt man sich nicht oft alleine. Hier kann es helfen mit anderen ins Gespräch zu kommen, die bereits in einer vergleichbaren Situation waren.
Sie möchten mit Betroffenen ins Gespräche kommen? Dann melden Sie sich gerne bei uns unter Wiebke.Roesler@usz.ch

Histiozytose. Und nun?
Unter histiozytäre Erkrankungen sind viele unterschiedliche Erkrankungen zusammengefasst. Am häufigsten treten im Erwachsenenalter die Langerhanszell-Histiozytose (LCH), die Erdheim-Chester-Erkrankung (ECD) und die Rosai-Dorfman-Destombes-Erkrankung (RDD) auf.
Histiozytose. Und nun?
Unter histiozytäre Erkrankungen sind viele unterschiedliche Erkrankungen zusammengefasst. Am häufigsten treten im Erwachsenenalter die Langerhanszell-Histiozytose (LCH), die Erdheim-Chester-Erkrankung (ECD) und die Rosai-Dorfman-Disease (RDD) auf.
Lange Zeit ist man davon ausgegangen, dass es sich hier um Erkrankungen handelt, denen eine Entzündung zu Grunde liegt.
Inzwischen weiss man aber, dass bei diesen drei Formen Veränderungen in bestimmten Blutzellen vorliegen, vor allem Zellen, die zur Immunabwehr gehören. Diese Veränderungen nennt man in der Regel «Mutationen», da es sich hier um Abweichungen in unserem genetischen «Bauplan», der DNA, handelt. Daher geht man nun davon aus, dass diese kranken Zellen ihre Umgebung aus unterschiedlichen Gründen so beeinflussen, dass eine Entzündung entsteht. Da in der Regel Zellen betroffen sind, die für die Infektabwehr im Gewebe zuständig sind, können diese kranken Zellen und damit die Erkrankung selber auch prinzipiell in jedem Gewebe auftauchen. Das kann die Diagnose oft schwierig machen, da es die ganz typischen Beschwerden eigentlich nicht gibt.
Durch das neue Wissen um diese Veränderungen sind aber auch neue Therapiemöglichkeiten entstanden und die Prognose konnte deutlich verbessert werden.
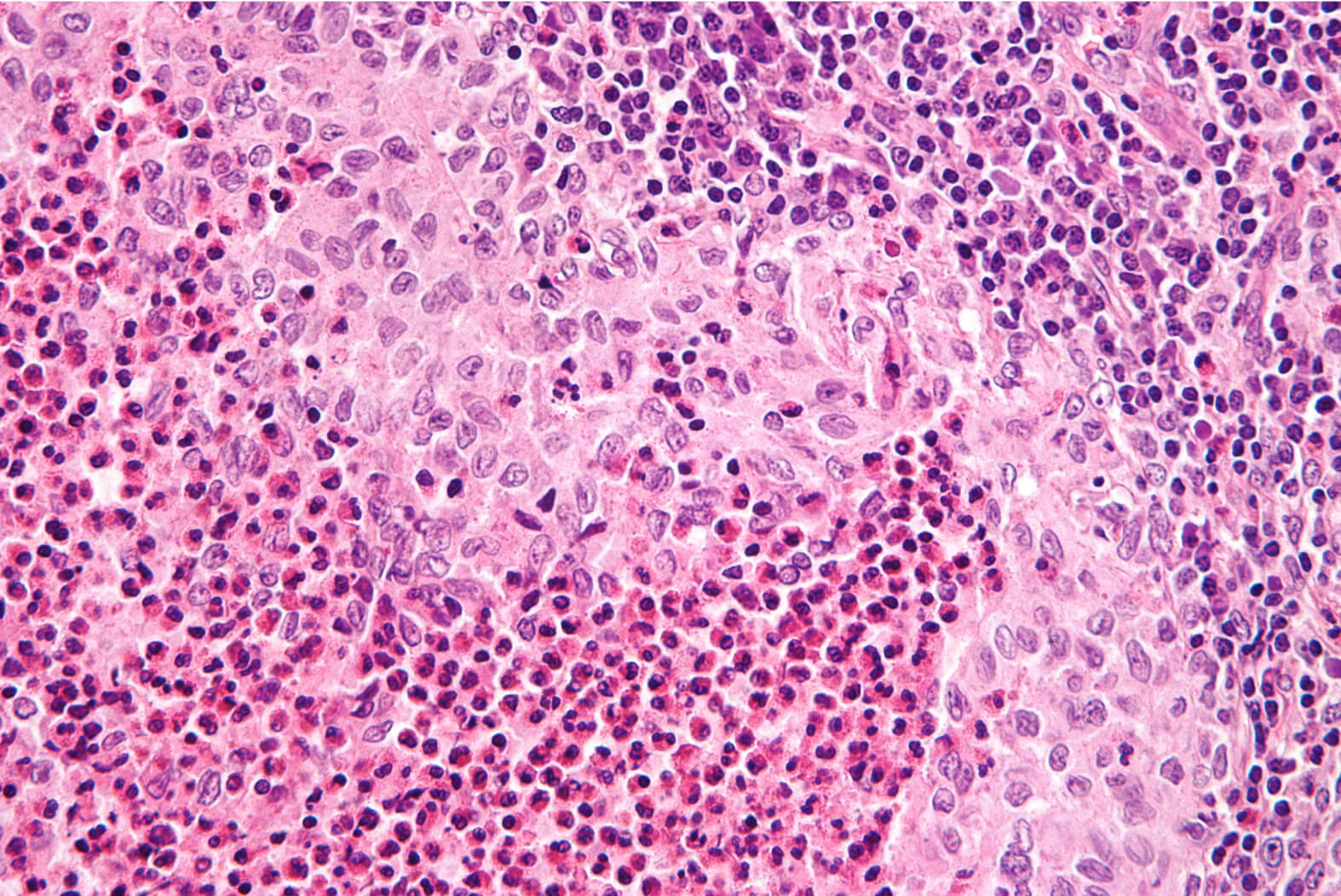
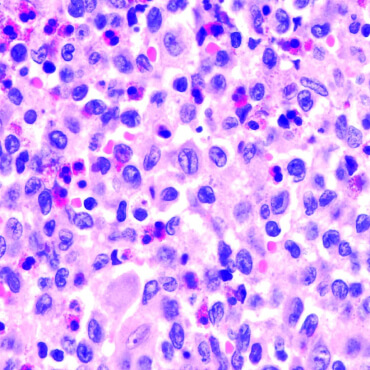
Langerhanszell-Histiozytose (LCH)
Die LCH ist eine seltene hämatologische Neoplasie, also eine Erkrankung des Blutbildenden Systems. Sie geht von Zellen aus, die später Abwehrzellen, vor allem im Gewebe bilden sollen.
Langerhanszell-Histiozytose (LCH)
Die LCH ist eine seltene hämatologische Neoplasie, also eine Erkrankung des Blutbildenden Systems. Sie geht von Zellen aus, die später Abwehrzellen, vor allem im Gewebe bilden sollen. Wenn man an LCH erkranktes Gewebe unter dem Mikroskop untersucht, findet man neben den erkrankten Zellen oft viele Entzündungszellen. Die erkrankten Zellen selber kann man durch unterschiedliche Färbemethoden und inzwischen auch genetische Untersuchungen erkennen und zuordnen.
Da diese Gewebsabwehrzellen in jedem Organ im Körper vertreten sind, kann die Erkrankung auch überall auftreten. Besonders häufig findet man sie aber in der Haut und in den Knochen, vor allem am Schädel. Soweit wir wissen, erkrankten von 1 Millionen Erwachsenen 1-2 Personen im Jahr daran. Es ist aber zu vermuten, dass es eine Dunkelziffer gibt, da die Erkrankung manchmal auch von alleine wieder weggehen kann. Das unterscheidet sie zum Beispiel auch von einer typischen Krebserkrankung.
Die Erkrankung ist nach allem, was wir im Moment wissen, nicht vererbbar. Als Risikofaktor ist vor allem bei einem Lungenbefall das Rauchen bekannt. Wenn man damit aufhört, bildet sich die Erkrankung in der Lunge oft wieder zurück.
Unterschiedliche Formen der LCH:
Grundsätzlich kann man die LCH in zwei grosse Gruppen unterteilen. Die eine Gruppe hat nur an einer einzelnen Stelle am Körper die Erkrankung. Dann reden wir von einer unifokalen single-system (ss)-LCH. Hier kann man je nach Ort gut eine lokale Behandlung machen und manchmal auch abwarten, ob überhaupt eine Behandlung notwendig ist. Es kann auch sein, dass ein Organsystem betroffen ist, aber an mehreren Stellen. Zum Beispiel nur Stellen am Knochen, aber dafür an mehreren unterschiedlichen Knochen. Oder an mehreren Stellen an der Haut. Dann sprechen wir von einer multifokalen single-system (ss-m) LCH.
Wenn unterschiedliche Organe betroffen sind, liegt eine multi-system (ms) LCH vor. Dann ist oft auch eine systemische Therapie, notwendig.
Diagnostik
Oft wird die LCH über eine Gewebeprobe festgestellt, wenn an einer bestimmten Stelle am Körper Beschwerden oder Veränderungen bestehen. Bei der ersten Vorstellung nach der Diagnose wird eine umfassende Untersuchung veranlasst. Neben der gründlichen Anamnese und körperlichen Untersuchung wird auch Blut abgenommen, um nach weiteren Hinweisen für betroffene Organe zu suchen (Leber, Niere, Blutbild, Hormone).
Ebenso ist nach der Diagnose eine Bildgebung vom gesamten Körper notwendig, ein sogenanntes PET-CT.
Gelegentlich wird gezielt das Gehirn mit einem MRT untersucht.
Zu einer kompletten Untersuchung gehört auch die Vorstellung bei den Hautärzten (Dermatologie) und bei den Augenärzten (Opthalmologie), um eine Befall der Haut und der Augen sicher zu finden oder ausschliessen zu können.
Weitere Untersuchungen, z.B. bei den Hormonspezialisten (Endokrinologen) oder Lungenärzten (Pneumologen) können notwendig sein. Dazu kann auch die Durchführung einer Lungenuntersuchung (Spirometrie) gehören.
Wenn es im Blutbild Auffälligkeiten gibt, ist manchmal eine Untersuchung des blutbildenden Knochenmarks notwendig, um die LCH auch dort suchen zu können. Hierbei wird unter lokaler Betäubung mit einer kleinen Nadel etwas Gewebe und Blut direkt aus dem Knochen (in aller Regel aus dem Beckenknochen am Rücken) entnommen. Der Eingriff wird ambulant durchgeführt und dauert in der Regel nur wenige Minuten. Anschliessend liegt man für circa 30 Minuten, um eine Blutung aus der Einstichstelle zu verhindern. Wenn dieser Eingriff notwendig ist, wird er vorher von den behandelnden Ärztinnen und Ärzten genau mit Ihnen besprochen werden.
Therapieformen
Die Therapie richtet sich nach dem Ausmass der Erkrankung und den Beschwerden.
Lokale Behandlungen
Operation:
Bei einzelnen Knochenherde wurde oft schon im Rahmen der Diagnostik das betroffene Gewebe entfernt. Dann muss gar nicht weiter behandelt werden. Fast 90% aller Patientinnen und Patienten haben danach nie wieder mit der Erkrankung zu tun. Auch einzelne Herde an der Haut können mit einem kleinen Eingriff entfernt werden.
Bestrahlung/Strahlentherapie:
Dies kommt vor allem bei einzelnen Knochenherden zum Einsatz. Wenn diese nicht mit einer Operation entfernt werden können, kann man diese Stellen mit einer Strahlentherapie behandeln. Auch hier sind die Behandlungsergebnisse sehr gut.
Immunsuppressive Behandlung:
Dies kommt vor allem bei der Haut zum Einsatz. Hier können Cremes, die Kortison oder andere das Immunsystem unterdrückende Substanzen enthalten, eingesetzt werden.
Kortison kann auch bei Knochenbefall angewendet werden. Dann wird eine kleine Menge Kortison direkt in die betroffene Stelle gespritzt.
Systemische Behandlungen
Hier gibt es eine Vielzahl von Medikamenten, die zum Einsatz kommen können. Man kann sie in folgende Gruppen einteilen:
Zytostatische Therapien/Chemotherapie:
Da die Erkrankung von blutbildenden Zellen ausgeht, versucht man mit diesen Therapien, diese Zellen zu zerstören. Es gibt unterschiedliche Substanzen, die zum Einsatz kommen können. Am häufigsten sind es die Medikamente Cladribin, Cytarabin oder Methotrexat, sowie Hydroxurea. Diese Medikamente zerstören die blutbildenden Zellen im Knochenmark, damit auch die erkrankten Zellen. Anschliessend sollen sich nur die gesunden Zellen wieder neu bilden. Je nach eingesetztem Medikament und Ansprechen auf die Therapie wird über 4-12 Monate behandelt. Am häufigsten tritt unter den Medikamenten eine Verschlechterung des Blutbildes auf, die aber nur kurzfristig ist. Trotzdem ist man in dieser Zeit gefährdetet für Infektionen und vielleicht auch müde und nicht so belastbar.
Meistens werden diese Medikamente als Infusion oder als Spritze unter die Haut gegeben. Methotrexat und Hydroxyurea können als Tabletten gegeben werden. Es kann auch sein, dass man diese Medikamente untereinander kombiniert.
Die Erfolgsraten sind hier sehr unterschiedlich, oft wurden nur wenige Patientinnen und Patienten behandelt, was die Aussage schwierig macht. Über alle genannten Substanzen erreicht man aber bei circa 50% der Patientinnen und Patienten eine komplette Rückbildung der Veränderungen. In anderen Fällen kann die Erkrankung zumindest gestoppt werden oder bildet sich teilweise zurück. Auch das kann ein gutes Behandlungsergebnis sein.
«Gezielte» Therapiemassnahmen:
Man konnte nachweisen, dass bei der LCH ein Fehler im Bauplan der Zellen vorliegt. Das führt dazu, dass Signalwege, die die Zellen benutzen, um bestimmten Informationen zu verarbeiten (zum Beispiel für die Zellteilung), zu viele Signale geben und die Zellen nicht mehr von alleine abstirbt, wenn es eigentlich notwendig wäre.
Oft liegen sogenannte «BRAF» oder «MEK»-Mutationen vor. Hierfür gibt es Medikamente, die gezielt diese überaktivierten Signalwege unterbrechen, die «BRAF»- oder «MEK»- Inhibitoren (Blocker). Hierbei handelt es sich um Tabletten. Welche Substanz zum Einsatz kommt, hängt von den spezifischen Veränderungen ab, ebenso von anderen Begleiterkrankungen und den möglichen Nebenwirkungen.
Die Therapien sind noch nicht lange im Einsatz. Zurzeit gehen wir aber davon aus, dass es Dauertherapien sind, denn man hat beobachtet, dass die LCH in der Regel wiederkommt, wenn man die Therapie beendet.
Begleittherapie:
Wenn die Knochen betroffen sind, empfehlt man eine zusätzliche Infusion, die die Knochen stärken soll (Bisphosphonate). Diese wird am Anfang alle 4 Wochen gegeben, nach einem Jahr kann die Gabe auf alle 3 Monate verlängert werden.
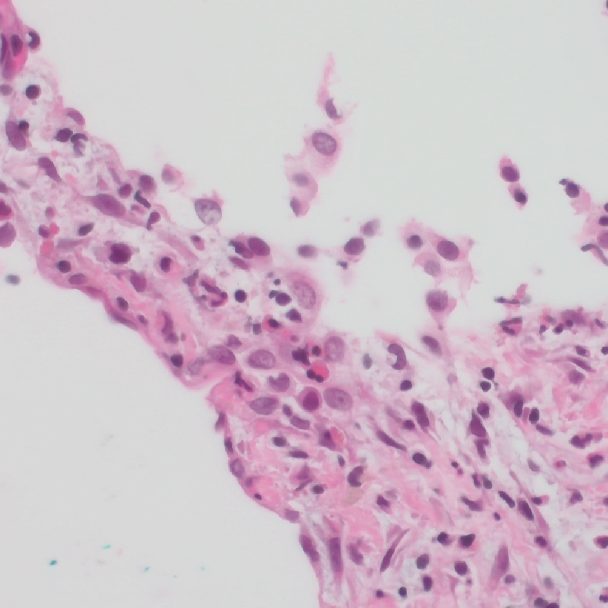
Erdheim-Chester-Erkrankung (Erdheim-Chester-Disease/ECD)
Auch die ECD gehört zu den selten histiozytären Erkrankungen. Sie ist noch seltener als die LCH, weltweit sind nur wenige Tausend Fälle beschrieben. Ähnlich der LCH entsteht sie aus Abwehrzellen.
Erdheim-Chester-Erkrankung (Erdheim-Chester-Disease/ECD)
Auch die ECD gehört zu den selten histiozytären Erkrankungen. Sie ist noch seltener als die LCH, weltweit sind nur wenige Tausend Fälle beschrieben. Ähnlich der LCH entsteht sie aus Abwehrzellen. Anders als die LCH tritt die ECD aber so gut wie nie lokalisiert, das heisst nur an einem Ort am Körper auf. Ebenso findet man die ECD fast ausschliesslich bei Erwachsenen. Meistens beginnt die Erkrankung im Alter zwischen 40 und 60 Jahren. Männer sind häufiger betroffen als Frauen.
Die ECD ist wie ein Chamäleon und kann sehr unterschiedliche Beschwerden auslösen, die oft und lange erst nicht richtig zugeordnet werden können. Häufig beklagen Betroffene Schmerzen in den Beinen, Müdigkeit und eine Verschlechterung der Stimmung bis zu psychischen Problemen (meist Depressionen/depressive Verstimmungen) oder Konzentrationsproblemen. Wenn der Hormonhaushalt betroffen ist, kann es sein, dass man sehr viel Durst hat und auch viel öfter Wasser lösen muss. Husten und Luftnot sind weitere Beschwerden. Die ECD führt häufig dazu, dass sich im Körper Bindegewebe bildet, wo es eigentlich nicht hingehört. Daher kann sie auch die Gefässe betreffen und sich wie ein Verschluss der Gefässe zeigen. Das kann zu Durchblutungsstörungen führen. Auch die Nieren können auf diese Weise betroffen sein. Hier gibt es ein sehr typisches Bild, das sogenannte «Hairy-Kidney-Sign.» Wenn man ein Bild der Nieren macht, sieht es aus als ob die Nieren Haare hätten.
Oft braucht es für die Diagnose alle Aspekte, also eine Probe aus dem betroffenen Gewebe, die klinischen Beschwerde und die Veränderung, die man in der sogenannten bildgebenden Diagnostik (Röntgenbild, CT, MRT) sieht.
Wie die LCH geht man im Moment nicht davon aus, dass die ECD vererbbar ist. Was die Erkrankung auslöst ist nicht bekannt. Allerdings findet man auch hier typische Veränderungen im Bauplan der Zellen, die sogenannten Mutationen. Das hat neue Therapiemöglichkeiten eröffnet.
Diagnostik der ECD
Oft ist die Diagnose der ECD über eine Gewebeprobe schwierig, da man nur bindegewebige und entzündliche Veränderungen sieht und nicht die kranken Zellen. Dann braucht man die Kombination aus den klinischen Beschwerden, der Bildgebung, die oft typische Veränderungen zeigt und der Gewebeprobe. Bei dem Verdacht auf eine ECD sind daher umfassende Untersuchungen notwendig.
Neben der gründlichen Anamnese und körperlichen Untersuchung wird Blut abgenommen, um nach weiteren Hinweisen für betroffene Organe zu suchen (Leber, Niere, Blutbild, Gefässe, Herz, Hormone).
Ebenso ist nach der Diagnose eine Bildgebung vom gesamten Körper notwendig, ein sogenanntes PET-CT. Hierbei ist es wichtig, dass wirklich der ganze Körper untersucht wird, da man typische Veränderungen der ECD oft auch in den Knochen der Unterschenkel sieht.
Auch wird in der Regel gezielt das Gehirn und das Herz mit einem MRT untersucht.
Zu einer kompletten Untersuchung gehört auch die Vorstellung bei den Hautärzten (Dermatologie) und bei den Augenärzten (Opthalmologie), um eine Befall der Haut und der Augen sicher zu finden oder ausschliessen zu können.
Weitere Untersuchungen, z.B. bei den Hormonspezialisten (Endokrinologen) oder Lungenärzten (Pneumologen) können notwendig sein. Dazu kann auch die Durchführung einer Lungenuntersuchung (Spirometrie) gehören.
Wenn es im Blutbild Auffälligkeiten gibt, ist manchmal eine Untersuchung des blutbildenden Knochenmarks notwendig, um die ECD auch dort suchen zu können. Hierbei wird unter lokaler Betäubung mit einer kleinen Nadel etwas Gewebe und Blut direkt aus dem Knochen (in aller Regel aus dem Beckenknochen am Rücken) entnommen. Der Eingriff wird ambulant durchgeführt und dauert in der Regel nur wenige Minuten. Anschliessend liegt man für circa 30 Minuten, um eine Blutung aus der Einstichstelle zu verhindern. Wenn dieser Eingriff notwendig ist, wird er vorher von den behandelnden Ärztinnen und Ärzten genau mit Ihnen besprochen werden.
Therapie der ECD
Die Therapie der ECD umfasst in der Regel den ganzen Körper, das heisst man gibt Tabletten, Spritzen oder Infusionen.
Die Therapie der ECD hat sich aber in den letzten Jahren dramatisch verändert.
Man konnte nachweisen, dass bei der ECD ein Fehler im Bauplan der Zellen vorliegt. Das führt dazu, dass Signalwege, die die Zellen benutzen, um bestimmten Informationen zu verarbeiten (zum Beispiel für die Zellteilung), zu viele Signale geben und die Zellen nicht mehr von alleine abstirbt, wenn es eigentlich notwendig wäre.
Oft liegen sogenannte «BRAF» oder «MEK»-Mutationen vor. Hierfür gibt es Medikamente, die gezielt diese überaktivierten Signalwege unterbrechen, die «BRAF»- oder «MEK»- Inhibitoren (Blocker). Hierbei handelt es sich um Tabletten. Welche Substanz zum Einsatz kommt, hängt von den spezifischen Veränderungen ab, ebenso von anderen Begleiterkrankungen und den möglichen Nebenwirkungen.
Die Therapien sind noch nicht lange im Einsatz. Zurzeit gehen wir aber davon aus, dass es Dauertherapien sind, denn man hat beobachtet, dass die ECD in der Regel wiederkommt, wenn man die Therapie beendet. Das Ansprechen auf die Therapie ist aber sehr gut und die möglichen Nebenwirkungen können wir inzwischen gut kontrollieren und behandeln, so dass die Betroffenen auch mit den Medikamenten eine sehr gute Lebensqualität haben.
Wenn diese Therapien nicht zum Einsatz kommen können oder nicht gewünscht werden, gibt es viele unterschiedliche Substanzen, die vor allem gegen die Entzündung wirken, die die ECD im Gewebe auslöst. Damit kann ebenfalls eine gute Kontrolle der Beschwerden erreicht werden.
Welche Therapie zum Einsatz kommt, wird der behandelnde Arzt/die behandelnde Ärztin detailliert mit den Betroffenen besprechen.
Lokale Therapie spielen bei der ECD in der Regel kaum eine Rolle. Auf eine Bestrahlung spricht die Erkrankung in der Regel nicht an. Sie ist daher nicht empfohlen. Eine Ausnahme gibt es, wenn schnell eine lokale Kontrolle von Beschwerden notwendig ist, zum Beispiel wenn wichtige Organe von der Erkrankung «eingedrückt» werden. Damit wird die Erkrankung aber nicht geheilt.
Prognose der ECD
Durch die neuen Medikamente, die sogenannten Inhibitoren, hat sich die Prognose für die ECD-Patientinnen und Patienten deutlich verbessert. Viele leben mit dieser Therapie viele Jahre mit guter Lebensqualität. Da diese Therapien aber noch nicht so lange eingesetzt werden, kann man zu der langfristigen Therapie noch wenig sagen.
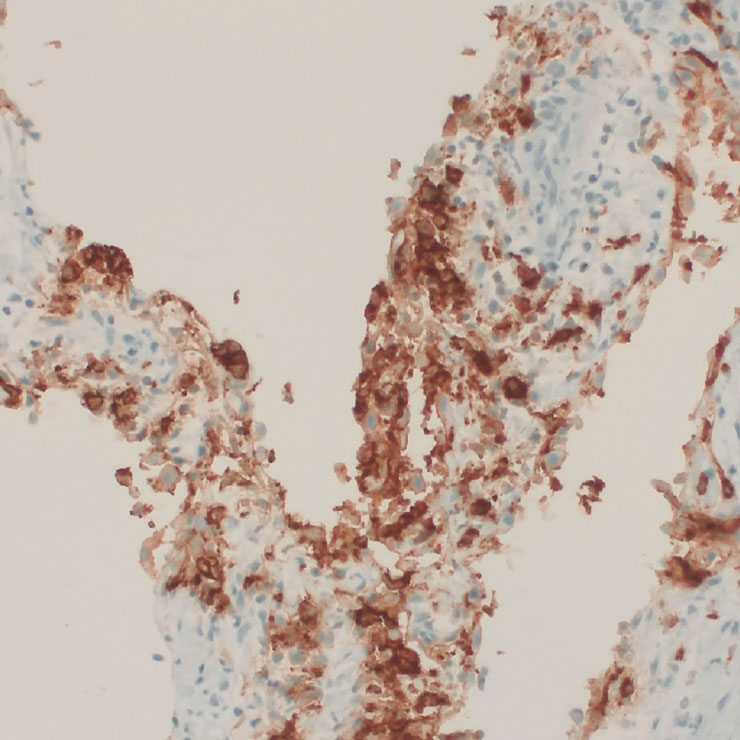
Rosai-Dorfman-Destombes Erkrankung (RDD)
Die Rosaï-Dorfman-Destombes Erkrankung ist eine seltene benigne Nicht-Langerhanszell-Histiozytose mit der Entwicklung großer schmerzloser histiozytärer Tumoren in den Lymphknoten vor allem der Halsregion
Rosai-Dorfman-Destombes-Erkrankung (Rosai-Dorfman- Destombes Disease, RDD)
Die RDD ist eine sehr seltene sogenannte Nicht-Langerhanszell-Histiozytose. Die Prävalenz beträgt 1:200.000, das heisst, von 200.000 Menschen hat einer oder eine RDD. Sie ist häufiger bei Kinder, Jugendlichen und jungen Erwachsenen, kann aber auch im höheren Alter vorkommen.
Die RDD zeichnet sich durch eine Ansammlung von bestimmten weissen Blutkörperchen aus, den sogenannten Histiozyten. Das sind Abwehrzellen, die vor allem im Gewebe ihre Arbeit verrichten. Die RDD kann in vielen unterschiedlichen Formen und klinische Zuständen auftreten. Sie kann alleine auftreten, oder im Rahmen von Autoimmun-Erkrankungen oder auch Krebserkrankungen. Deswegen sollte man wenn eine RDD diagnostiziert wurde nach anderen Erkrankungen suchen.
Auch bei der RDD hat man inzwischen festgestellt, dass Mutationen, also Veränderungen im Bauplan einer Zelle, bei der Erkrankung eine Rolle spielen können. Aber man weiss hier noch viel weniger als bei der ECD und der LCH. Die RDD kann in zwei grosse Gruppen aufgeteilt werden. Die klassische (nodale) RDD, welche die Lympknoten (daher Nodulus/nodal) betrifft und die extranodale RDD (also ausserhalb der Lymphknoten).
Bei der nodalen RDD zeigt sich in der Regel an beiden Seiten am Hals grosse Schwellungen der Lymphknoten, welche aber nicht schmerzhaft sind. Dazu können Fieber, Nachtschweiss und Gewichtsverlust auftreten. Das ist aber nicht bei allen Betroffenen der Fall. Die extranodale RDD kann fast alle Organe betreffen. Relativ häufig findet man die Erkrankung dann an der Haut, an Nase und Nasennebenhöhlen, im Gehirn und Nervensystem, sowie an den Augen und Knochen. Es finden auch Veränderungen im Blutbild, allerdings ist das Knochenmark, wo das Blut gebildet wird nur sehr selten auch erkrankt. Die Beschwerden richten sich dann nach dem Organ, dass betroffen ist.
Es gibt eine seltene Form einer familiären RDD. In anderen Fällen gehen wir derzeit nicht davon aus, dass die Erkrankung vererbt werden kann. Risikofaktoren im eigentlichen Sinn sind nicht bekannt. Es wird aber diskutiert, ob neben Autoimmunerkrankungen und Krebserkrankungen auch Virusinfekte die Erkrankung auslösen können.
Diagnostik der RDD
In der Regel wird die RDD über eine Gewebeprobe festgestellt, wenn an einer bestimmten Stelle am Körper Beschwerden oder Veränderungen bestehen. Bei der ersten Vorstellung nach der Diagnose wird eine umfassende Untersuchung veranlasst. Neben der gründlichen Anamnese und körperlichen Untersuchung wird auch Blut abgenommen, um nach weiteren Hinweisen für betroffene Organe (Leber, Niere, Blutbild, Hormone) oder Autoimmunerkrankungen zu suchen.
Ebenso ist nach der Diagnose eine Bildgebung vom gesamten Körper notwendig, das kann ein sogenanntes PET-CT sein, es kommen aber auch andere Methoden in Frage (Ultraschall, Röntgenbilder oder ein MRT). Gelegentlich wird gezielt das Gehirn mit einem MRT untersucht. Weitere Untersuchungen, wie z.B. eine Untersuchung der Lungenfunktion, können notwendig sein.
Wenn es im Blutbild Auffälligkeiten gibt, ist manchmal eine Untersuchung des blutbildenden Knochenmarks notwendig, um die RDD auch dort zu suchen. Hierbei wird unter lokaler Betäubung mit einer kleinen Nadel etwas Gewebe und Blut direkt aus dem Knochen (in aller Regel aus dem Beckenknochen am Rücken) entnommen. Der Eingriff wird ambulant durchgeführt und dauert in der Regel nur wenige Minuten. Anschliessend liegt man für circa 30 Minuten, um eine Blutung aus der Einstichstelle zu verhindern. Wenn dieser Eingriff notwendig ist, wird er vorher von den behandelnden Ärztinnen und Ärzten genau mit Ihnen besprochen werden.
Ebenso kann eine Untersuchung des Liquors (Flüssigkeit, die das Gehirn und das Rückenmark umgibt) notwendig sein. Dann wird mit einer sehr dünnen Nadel weit unten an der Wirbelsäule etwas von diesem Rückenmarkswasser entnommen. Anschliessend muss man wie bei der Knochenmarkspunktion eine bestimmte Zeit flachliegen. Damit sollen vor allem Kopfschmerzen, wie sie nach einer solchen Punktion auftreten können, vermindert werden. Auch dieser Eingriff wird er vorher von den behandelnden Ärztinnen und Ärzten genau mit Ihnen besprochen werden.
Therapie der RDD
Für die Therapie der RDD gibt es keine festgelegte Therapie, sondern in sie wird sehr individuell auf die klinischen Beschwerden der Betroffenen abgestimmt.
Wenn die Erkrankung keine Beschwerden macht, dann kann einfach abgewartet werden. Gerade bei Betroffenen, die nur einen Befall der Lymphknoten oder der Haut haben, bildet sich die Erkrankung in bis zur Hälfte der Fälle zurück.
Lokale Behandlungen:
Operation:
Wenn nur einzelne Stellen betroffen sind, können diese mittels einer Operation entfernt werden, das gilt vor allem für die Haut. Hier ist das sogar die effektivste Therapiemöglichkeit.
Bestrahlung/Strahlentherapie:
Die Strahlentherapie verfügt bei der RDD nur über eine begrenzte Wirksamkeit, kann aber in Einzelfällen sinnvoll sein, vor allem, wenn die Erkrankung nach einer Operation an einer Stelle am Körper wiedergekommen ist.
Systemische Behandlungen:
Eine Systemtherapie ist dann notwendig, wenn die Erkrankung an unterschiedlichen Stellen am Körper sitzt und/oder dadurch Beschwerden bestehen, die zum Beispiel durch eine Operation nicht gelindert werden können.
Für eine solche Systemtherapie gibt es ganz unterschiedliche Möglichkeiten. Hier muss man wissen, dass die Medikamente, die zum Einsatz kommen, für die RDD in der Regel nicht zugelassen sind. Es gibt aber ausreichende wissenschaftliche Berichte und Erfahrungen dazu. Dies werden die behandelnden Ärztinnen und Ärzte mit Ihnen ausführlich besprechen.
Da oft nur einzelne Fälle für die unterschiedlichen Therapiemöglichkeiten mit auch sehr unterschiedlichen Ergebnissen beschrieben sind, gehen wir hier nicht auf alle Medikamente ein. Grundsätzlich kann man sagen, dass neben Kortison auch andere, das Immunsystem unterdrückende Substanzen zum Einsatz kommen können. Gelegentlich kann auch eine Chemotherapie notwendig sein. Wenn bestimmte Veränderungen im Bauplan einer Zelle vorliegen (Mutation), dann kann auch eine «gezielte» Therapie zum Einsatz kommen, die sich genau gegen die Veränderungen richtet.
Prognose der RDD
Es gibt auch hier nur sehr wenige Daten. Wenn nur die Lymphknoten oder die Haut betroffen ist, dann ist der Verlauf oft sehr gut, die Erkrankung kann dann auch von alleine zurückgehen oder ist mit einer Entfernung der erkrankten Stelle durch eine Operation geheilt. Wenn die Nieren, die Leber oder die Lungen betroffen sind, braucht man oft eine intensivere Therapie. Aber auch hier haben > 90% der Betroffenen dann ein gutes Ansprechen und einen zufriedenstellenden Verlauf.
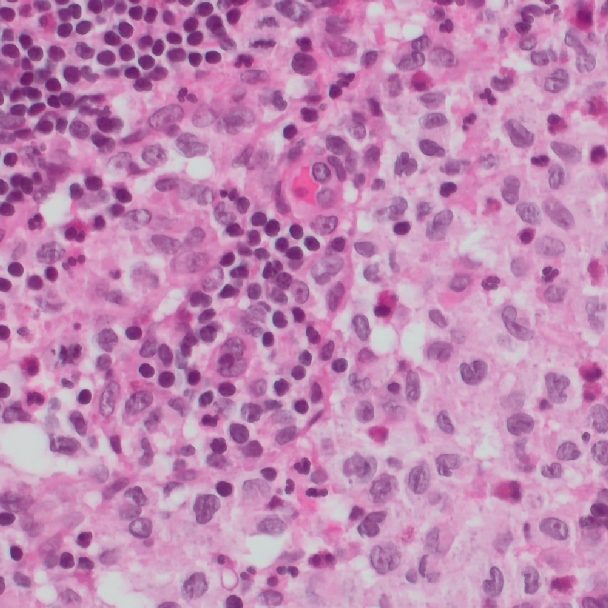
Klinische Studien
Registerstudien Universitätsspital Zürich
Seit 2020 gibt es an der Klinik für Medizinische Onkologie und Hämatologie am Universitätsspital Zürich eine Registerstudie. Hier sammeln wir alle Daten, die im Rahmen der normalen Untersuchungen und Therapien erhoben werden.
Das Histiozytose Register (Registerstudie)
Histiozytäre Erkrankung sind sehr selten. Oftmals lassen sich Erkrankungen aus diesem Formenkreis zwar heute bereits besser behandeln als noch vor einigen Jahren, aber es gibt noch immer Bedarf zur weiteren Therapieoptimierung und zahlreiche offene Fragen: Mit welchen Massnahmen lässt sich der Verlauf der Erkrankung am besten beeinflussen? Was treten bei einer Therapie für Nebenwirkungen auf und wie lassen sich diese vermeiden oder lindern? Wie beeinflussen Krankheit und Therapie Ihre Lebensqualität? Wie ist der Langzeitverlauf dieser Erkrankung? Um solche Fragen besser untersuchen und damit betroffenen Patientinnen und Patienten noch gezielter helfen zu können, haben wir eine Registerstudie am Universitätsspital initiiert. Wenn Sie hierzu mehr wissen oder an dem Register als Betroffene/Betroffener oder Behandlerin/Behandler teilnehmen möchten, wenn Sie sich bitte an: Wiebke.Roesler@usz.ch
Veranstaltungen
1. Schweizer Histiozytose-Symposium
Am 20.10.2023 trafen sich Expertinnen und Experten zum 1. Schweizer Histiozytose-Symposium am Universitätsspital Zürich. Begrüßt wurden die Teilnehmerinnen und Teilnehmer von Prof. Markus G. Manz, Klinikdirektor der Klinik für Medizinische Onkologie und Hämatologie. Er verwies angesichts der Seltenheit histiozytärer Erkrankungen einerseits sowie dem hohen Leidensdruck der Betroffenen andererseits auf den dringenden Bedarf nicht nur an […]
1. Schweizer Histiozytose-Symposium
Am 20.10.2023 trafen sich Expertinnen und Experten zum 1. Schweizer Histiozytose-Symposium am Universitätsspital Zürich. Begrüßt wurden die Teilnehmerinnen und Teilnehmer von Prof. Markus G. Manz, Klinikdirektor der Klinik für Medizinische Onkologie und Hämatologie. Er verwies angesichts der Seltenheit histiozytärer Erkrankungen einerseits sowie dem hohen Leidensdruck der Betroffenen andererseits auf den dringenden Bedarf nicht nur an […]
Downloadbereich
Ansprechpartner
Universitätsspital Zürich
Dr. med. Wiebke Rösler, Oberärztin
Klinik für Medizinische Onkologie und Hämatologie
Rämistrasse 100
8091 Zürich, Schweiz
Kontakt:
+41 44 255 11 11
wiebke.roesler@usz.ch
Lausanne, CHUV
Dr. Grégoire Stalder, Médecin agréé
Service et laboratoire central d’hématologie, Centre Hospitalier Universitaire Vaudois (CHUV)
Rue du Bugnon 46
1011 Lausanne, Schweiz
Kontakt:
+4127 603 67 59
gregoire.stalder@chuv.ch
Bern, Inselspital
Prof. Dr. med. Alicia Rovó, Stv. Klinikdirektorin Med. Bereich und Stv. Chefärztin Universitätsklinik für Hämatologie und Hämatologisches Zentrallabor
Freiburgstrasse, BHH A1456
3010 Bern, Schweiz
Kontakt:
+4131 632 3313
Alicia.Rovo@insel.ch
Winterthur, Kantonspital
Dr. med. Martina Bertschinger, Oberärztin Klinik für Medizinische Onkologie und Hämatologie
Brauerstrasse 15
8401 Winterthur, Schweiz
Kontakt:
+4152 266 21 21
martina.bertschinger@ksw.ch
St. Gallen, Kantonspital
Prof. Dr. med. Markus Schittenhelm, Leitender Arzt / Ärztliche Leitung Ambulatorium Spital Linth, Klinik für Medizinische Onkologie und Hämatologie
Rorschacher Strasse 95
9007 St. Gallen, Schweiz
Kontakt:
+4171 494 29 22
marcus.schittenhelm@kssg.ch
Bellinzona, EOC
PD Dr.med. Bernhard Gerber, Klinik für Hämatologie,Ente Ospedaliero Cantonale
Viale Officina 3
6500 Bellinzona, Schweiz
Kontakt:
+4191 181 186 65
ematologica.ticino@eoc.ch
Institut Central des Hôpitaux, Sion
Dr. Grégoire Stalder, Médecin-adjoint, Service d’hématologie, Institut Central des Hôpitaux (ICH)
Av. Grand-Champsec 86
1950 Sion, Schweiz
Universitätsspital Zürich
Dr. med. Wiebke Rösler, Oberärztin
Klinik für Medizinische Onkologie und Hämatologie
Rämistrasse 100
8091 Zürich, Schweiz
Kontakt:
+41 44 255 11 11
wiebke.roesler@usz.ch
Lausanne, CHUV
Dr. Grégoire Stalder, Médecin agréé
Service et laboratoire central d’hématologie, Centre Hospitalier Universitaire Vaudois (CHUV)
Rue du Bugnon 46
1011 Lausanne, Schweiz
Kontakt:
+4127 603 67 59
gregoire.stalder@chuv.ch
Bern, Inselspital
Prof. Dr. med. Alicia Rovó, Stv. Klinikdirektorin Med. Bereich und Stv. Chefärztin Universitätsklinik für Hämatologie und Hämatologisches Zentrallabor
Freiburgstrasse, BHH A1456
3010 Bern, Schweiz
Kontakt:
+4131 632 3313
Alicia.Rovo@insel.ch
Winterthur, Kantonspital
Dr. med. Martina Bertschinger, Oberärztin Klinik für Medizinische Onkologie und Hämatologie
Brauerstrasse 15
8401 Winterthur, Schweiz
Kontakt:
+4152 266 21 21
martina.bertschinger@ksw.ch
St. Gallen, Kantonspital
Prof. Dr. med. Markus Schittenhelm, Leitender Arzt / Ärztliche Leitung Ambulatorium Spital Linth, Klinik für Medizinische Onkologie und Hämatologie
Rorschacher Strasse 95
9007 St. Gallen, Schweiz
Kontakt:
+4171 494 29 22
marcus.schittenhelm@kssg.ch
Bellinzona, EOC
PD Dr.med. Bernhard Gerber, Klinik für Hämatologie,Ente Ospedaliero Cantonale
Viale Officina 3
6500 Bellinzona, Schweiz
Kontakt:
+4191 181 186 65
ematologica.ticino@eoc.ch
Institut Central des Hôpitaux, Sion
Dr. Grégoire Stalder, Médecin-adjoint, Service d’hématologie, Institut Central des Hôpitaux (ICH)
Av. Grand-Champsec 86
1950 Sion, Schweiz